Data Core:
The Harvard PRECISION Human Pain Center proposes to use human TG/DRG tissue and painful neuromas to generate large-scale multi- omics datasets for the discovery and characterization of functional genetic elements, epigenetic signatures, and molecular/cellular pathways that underlie human pain transduction, transmission, and processing. As one of the key components of the Center, the Data Core aims to act as a data center by building data infrastructure, providing data management, coordination, analysis, and sharing to the investigators within the Center and other centers in the community. The projects will generate nearly 4,000 datasets of clinical (pain scores, demographics), single-cell RNA-seq, single-cell ATAC-seq, SNP genotyping, spatial transcriptomics from MERFISH, and physiology for over 1,700 samples from 150 donors.
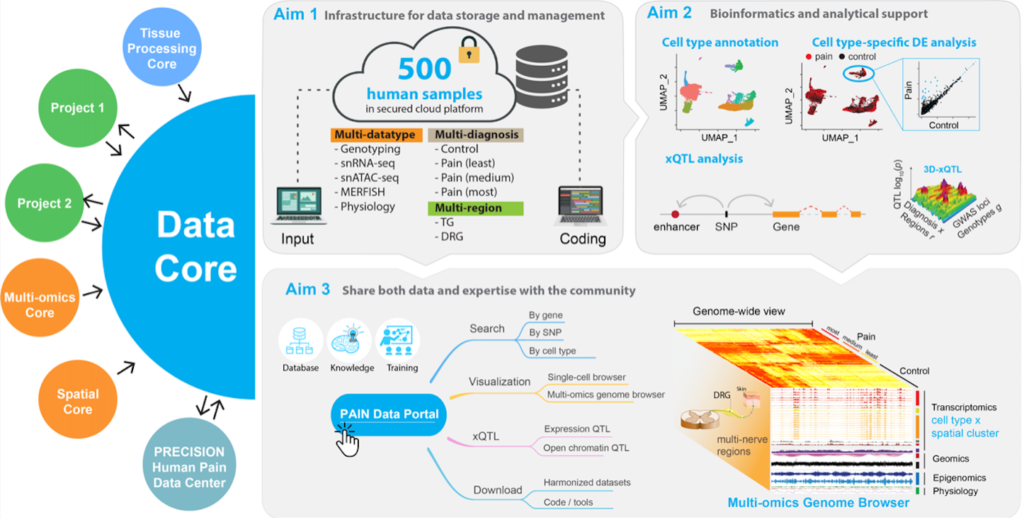
The Data Core will first harmonize the datasets and store in centralized MySQL database with accessible web interface for other cores in the Center. Standard bioinformatics pipelines will be developed to process the raw data in a systematic manner, including cell type annotation and spatial location in each brain region. The Data Core will also support the research Projects on their customized analysis, including power analysis, differential expression and chromatin accessibility across different cell types, brain regions, and diagnosis. It will also reveal the genetic variants that are associated with various molecular features via xQTL analysis. Last, all these computed blocks and tools will be integrated into a comprehensive, cloud-based, interactive web portal – Pain Data Portal, providing a generally useful resource for generating biological hypotheses, discovering effective diagnosis biomarkers, and developing new treatments for pain. The Data Core will also ensure a timely data transfer and expertise sharing with the other U19 and U24 centers in the community.
Spatial Core:

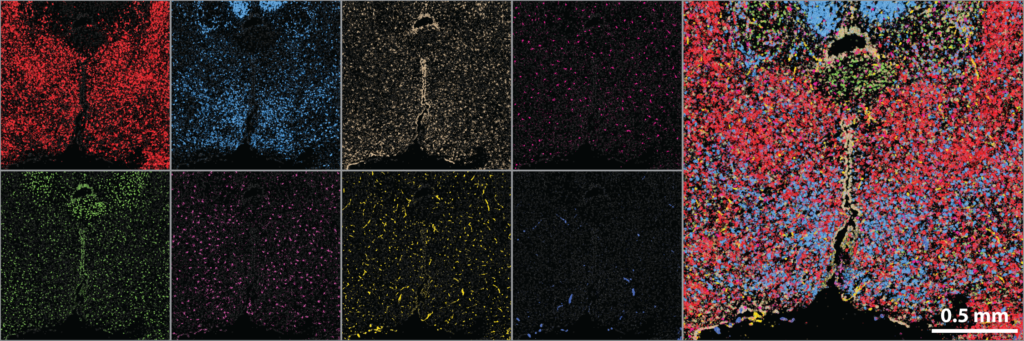
Image-based approaches to single-cell transcriptomics are the emerging and exciting answer to this demand, as these methods quantify single-cell expression profiles within fixed, intact samples via massively multiplexed, single-molecule RNA imaging. One such approach is MERFISH (multiplexed, error-robust, fluorescence in situ hybridization) which distinguishes individual RNAs with binary barcodes read out through sequential rounds of FISH staining and imaging. MERFISH has the proven ability to profile hundreds to ten thousand RNAs with near 100%- capture efficiency, single-molecule sensitivity, high throughput, and sub-micron resolution, and to leverage these capabilities to discover, define, and map cell types within the brain.
MERFISH was co-developed by Jeffrey Moffitt and is being extensively used and extended by the Moffitt laboratory. Thus, we have a unique opportunity to develop a spatial core in one of the few laboratories capable of these measurements and with the depth of expertise to help chart its use for the scientific goals of this center. Importantly, by integrating with a key laboratory in the spatial transcriptomics space, our core has access to the cutting edge of image-based transcriptomics, via MERFISH, and, critically, will continuously evaluate the emerging landscape of spatial profiling technologies, incorporating new technical capabilities as they emerge.